
ę╗Īó╗∙▒Šą┼Žó
Ė┼╩÷Ż║Angelman╩Ž░Y║“╚║Ż©╠ņ╩╣ŠC║Žš„Ż®Ż©Angelman syndromeŻ¼ASŻ®╩Ūę╗ĘNė╔ė┌─Ėį┤15q11-13╚Š╔½¾wģ^ė“Ą─UBE3A╗∙ę“▒Ē▀_«É│Ż╗“╣”─▄╚▒Ž▌ę²░lĄ─╔±Įø░lė²šŽĄKąį╝▓▓ĪĪŻų„ę¬▒Ē¼F×ķŠ½╔±░lė²▀t£■╗“ųŪ┴”Ą═Ž┬Ż¼šZčįĪó▀\äė╗“ŲĮ║Ō░lė²šŽĄKŻ¼┐ņśĘąą×ķŻ©╚ńŅlĘ▒░lą”Īó╬óą”╗“┼dŖ^Ż®Ż¼ąĪŅ^╗¹ą╬Ż¼░d░BĄ╚ĪŻ
▓Īę“Ż║─Ėį┤UBE3A╗∙ę“Ą─▒Ē▀_╗“╣”─▄╚▒Ž▌ī¦ų┬ASĪŻęč├„┤_Ą─Ęųūė▀zé„ÖCųŲėą4ĘNŻ║─Ėį┤15q11-13╚▒╩¦ĪóĖĖį┤15╠¢╚Š╔½¾w┤µį┌å╬ėHČ■¾wŻ©Uniparental disomyŻ¼ UPDŻ®Īó─Ėį┤15q11.2-q13ėĪėø╚▒Ž▌Ż©imprinting defectŻ¼ IDŻ®║═─Ėį┤UBE3A╗∙ę“░l╔·ų┬▓Ī═╗ūāĪŻ╦─ĘNĘųūėÖCųŲČ╝ī¦ų┬┴╦─Ėį┤UBE3A╗∙ę“Ą─╣”─▄å╩╩¦ĪŻŲõųą─Ėį┤15q11Ī½13╚▒╩¦ūŅ│ŻęŖŻ¼ČÓöĄ╝sķL5Ī½7 MbĪŻ
UBE3A╗∙ę“ŠÄ┤aĄ─Ę║╦žĄ░░ū▀BĮė├ĖE3Aģó┼c┴╦Ę║╦ž╗»═ŠÅĮŻ¼ī”╠žČ©Ą░░ū▀MąąĮĄĮŌĪŻį┌╚╦ŅÉ╠źā║─XĮM┐Ś║═│╔╚╦Ņ~╚~Ųż┘|ųąŻ¼UBE3A╗∙ę“ų„ę¬▒Ē¼F×ķ─Ėį┤▒Ē▀_Ż¼ĖĖį┤ę“╝ū╗∙╗»▓╗▒Ē▀_ĪŻš²│ŻŪķørŽ┬Ż¼╔±ĮøŽĄĮyė╔╣”─▄ąįĘ║╦ž-Ą░░ū├Ė¾wŽĄĮy▀MąąŲĮ║Ō╗“ŠS│ųŻ¼«öUBE3A╩¦╣”─▄Ģrät┐╔─▄ė░ĒæįōŽĄĮyŻ¼ę²Ų╗╝š▀║┌┘|Īó╝yĀŅ¾wĪó║Ż±R╝░ąĪ─XŲų┐Žę░╝Ü░¹Ą░░ūĘ║╦ž╗»«É│ŻĪŻ┴Ē═ŌŻ¼Ę║╦ž-Ą░░ū├Ė¾wŽĄĮyī”╝Ü░¹╣”─▄ę▓ų┴ĻPųžę¬Ż¼░³└©ą┼╠¢▐Dī¦Īó╝Ü░¹ų▄Ų┌▀M│╠ĪóDNAą▐Å═║═▐Dõøš{╣ØĪŻ╚╗Č°Ż¼įō╝▓▓Īī¦ų┬Ę║╦ž╗»«É│ŻĄ─Š▀¾w▓Ī└Ē╔·└ĒÖCųŲ╔ą▓╗═Ļ╚½ŪÕ│■ĪŻ
┴„ąą▓ĪīWŻ║ASį┌ÜW├└╚╦╚║╗╝▓Ī┬╩×ķ1/24000Ī½1/12000ĪŻ╬ęć°ČÓ×ķ╔ó░lł¾Ą└Ż¼╔ą¤oŽÓĻP┴„ąą▓ĪīWš{▓ķł¾ĖµĪŻ
Č■Īó╝▓▓Īį\öÓ
┼R┤▓▒Ē¼FŻ║╗╝š▀į┌ą┬╔·ā║Ų┌│Ż¤o«É│Ż▒Ē¼FĪŻ╩ū░l░YĀŅ╩Ū6į┬²gū¾ėę│÷¼F░lė²▀t£■▒Ē¼FŻ¼Ą½Ąõą═┼R┤▓▒Ē¼FČÓį┌1Üq║¾│÷¼FŻ¼┼R┤▓┤_Č©į\öÓ═∙═∙ąĶę¬öĄ─ĻĢrķgĪŻ░┤šš┼R┤▓▒Ē¼F│÷¼FŅl┬╩▓╗═¼Ęų×ķŻ║Š∙│÷¼FĄ─▒Ē¼FŻ©100Żź╗╝š▀Ż®ĪóĮø│Żąį▒Ē¼FŻ©80Żźęį╔Ž╗╝š▀Ż®ĪóŽÓĻPąį▒Ē¼FŻ©20ŻźĪ½80Żź╗╝š▀Ż®3ŅÉĪŻ
1.Š∙│÷¼FĄ─▒Ē¼F
š²│Żįą«a╩Ę║═│÷╔·Ņ^ć·Ż╗¤o│÷╔·╚▒Ž▌║═╔·╗»ųĖś╦«É│ŻŻ╗’B─XMRI/CT│²▌p╬óŲż┘|╬«┐sĪó╦ĶŪ╩░lė²▓╗┴╝═ŌŻ¼ČÓ¤oĮYśŗ«É│ŻŻ╗▀\äė└’│╠▒«┬õ║¾Č°¤oĄ╣═╦Ż╗6Ī½12éĆį┬│÷¼Fć└ųž░lė²▀tŠÅŻ╗šZčįšŽĄK’@╩Š×ķ¤o╗“śO╔┘┴┐į~ģRŻ¼ųžÅ═ąįšZčį║═ĘŪšZčįĮ╗═∙─▄┴”ÅŖė┌▒Ē▀_ąįšZčį─▄┴”Ż╗▀\äė╗“ŲĮ║ŌšŽĄK│Ż▒Ē¼F×ķ╣▓Ø·╩¦š{ęį╝░╦─ų½šŅØŻ╗«É│ŻĄ─ąą×ķ╠žš„▒Ē¼F×ķŅlĘ▒┤¾ą”╗“╬óą”Īó├„’@Ą─┼dŖ^äėū„╗“┐ņśĘ┼eų╣Īó│Ż░ķ┼─╩ų╗“ČÓäėĪŻ
2.Įø│Żąį▒Ē¼F
Ņ^ć·į÷ķL┬õ║¾Ż¼ļSįLų┴2Üq╚į▒Ē¼F×ķąĪŅ^╗¹ą╬Ż╗3ÜqŪ░│Ż│÷¼F░d░BŻ╗«É│Ż─XļŖłDŻ║╠žš„ąįĖ▀▓©Ę∙╝¼-┬²▓©ĪŻ
3.ŽÓĻPąį▒Ē¼F
šĒ▓┐▒ŌŲĮŻ╗Ž▓═┬╔ÓŻ╗╬³╦▒╗“═╠č╩šŽĄKŻ╗ŗļā║Ų┌╬╣B└¦ļyŻ╗╝ĪÅł┴”Ą═Ż╗Š▐┤¾Ž┬ŅMŻ╗č└ķgŽČīÆŻ╗ŅlĘ▒┴„ŽčŻ╗▀^Č╚ŠūĮ└äėū„Ż╗ą▒ęĢŻ╗Ųż─w╔½╦ž£p═╦Ż¼┼c╝ę╚╦ŽÓ▒╚Ņ^░l║═č█Š”Ņü╔½£\Ż©āHęŖė┌╚▒╩¦ą═Ż®Ż╗Ž┬ų½ļņĘ┤╔õ┐║▀MŻ╗╔Ž┼e╗“ÅØŪ·╔Žų½Ż¼ė╚Ųõ╩Ūąąū▀ĢrŻ╗▓Į╗∙īÆŻ╗ī”¤ß├¶ĖąąįĖ▀Ż╗«É│Ż╦»├▀-ėXąčų▄Ų┌Īó╦»├▀╔┘Ż╗├įæ┘╦«Īó╝łĪó╦▄┴ŽŻ╗▀M╩│ŽÓĻPąą×ķ«É│ŻŻ╗Ę╩┼ųŻ©ČÓęŖė┌─ĻķLĪóĘŪ╚▒╩¦ą═╗╝š▀Ż®Ż╗╝╣ų∙é╚═╣Ż╗▒Ń├žĪŻ
▌oų·Öz▓ķŻ║
1.▀zé„īWÖz▓ķ
▀zé„īWÖz▓ķ×ķįō▓Ī┤_į\╩ųČ╬ĪŻŠ▀¾w░³└©DNA╝ū╗∙╗»Ęų╬÷Ż¼▀@ŅÉĘĮĘ©░³└©╝ū╗∙╗»ČÓųž▀BĮėę└┘ć╩Į╠ĮßśöUį÷╝╝ągŻ©MS-MLPAŻ®Īó╝ū╗∙╗»PCRŻ©MS-PCRŻ®Ą╚Ż╗UBE3A╗∙ę“ą“┴ąĘų╬÷Ż╗SNP-arrayąŠŲ¼Ęų╬÷┐╔ęįÖz£y15╠¢╚Š╔½¾wĄ─╬ó╚▒╩¦║═UPDĪŻ
2.─XļŖłD
ASŠ▀ėąŽÓī”╠žš„ąįĄ─EEG«É│ŻŻ¼│Ż─▄į┌┼R┤▓░YĀŅ├„’@Ū░╝░╗∙ę“į\öÓŪ░╠ß╩Š▒Š▓ĪŻ¼Å─Č°ėąų·ė┌įńŲ┌į\öÓĪŻ─┐Ū░▒╚▌^╣½šJĄ─AS╠žš„ąį«É│ŻEEG░³└©3ĘNłDą╬Ż║
Ż©1Ż®”─łDą╬Ż╗
Ż©2Ż®”╚łDą╬Ż╗
Ż©3Ż®║¾Ņ^▓┐╝¼┬²▓©łDą╬ĪŻ
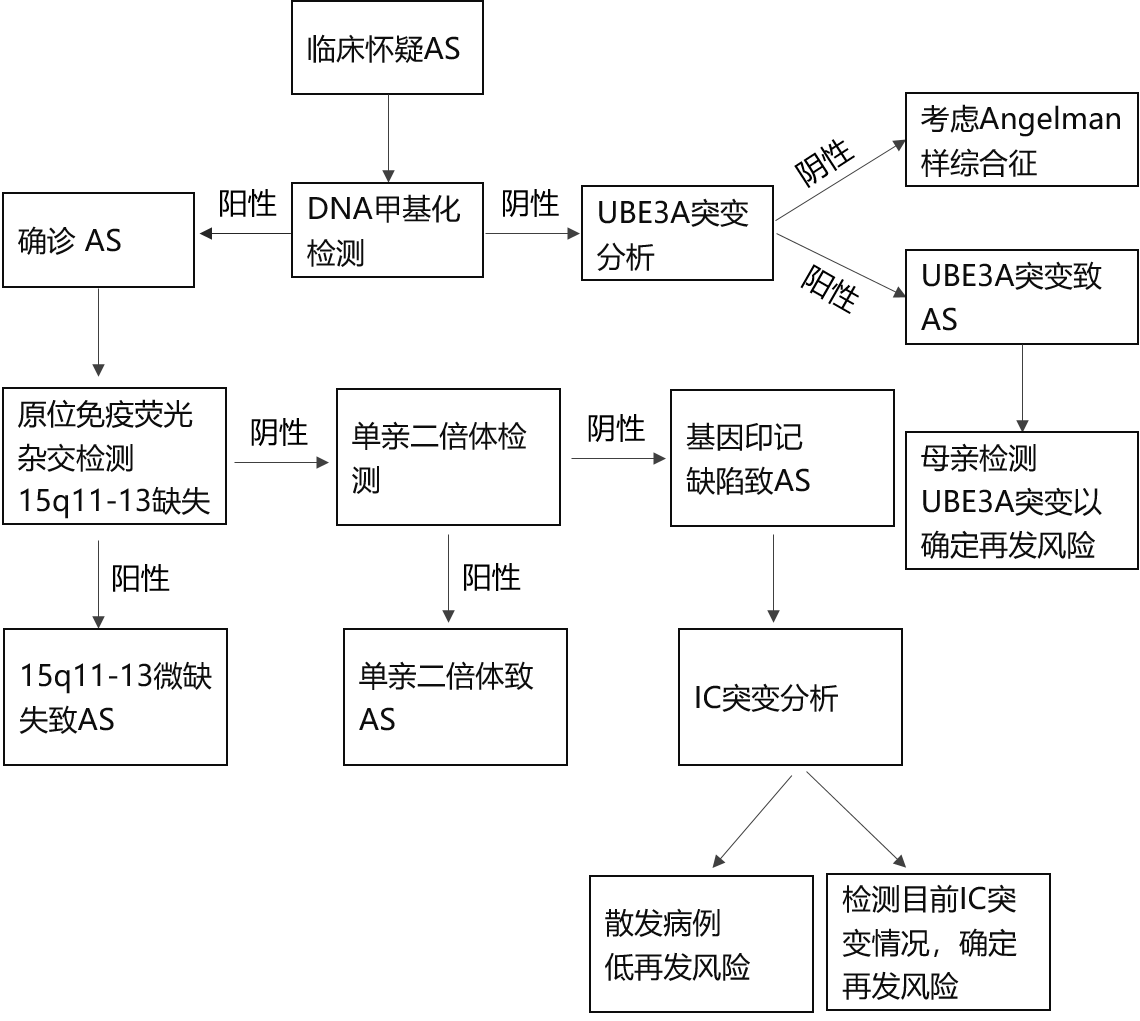
į\öÓŻ║Ę¹║ŽAS┼R┤▓į\öÓś╦£╩╣▓ūR║═Ż©╗“Ż®Ęųūė▀zé„īWÖz£yĮY╣¹▒Ē├„─Ėį┤UBE3AĄ╚╬╗╗∙ę“┤µį┌▒Ē▀_╗“╣”─▄╚▒Ž▌ĢrŻ¼╝┤┐╔ĮoėĶį\öÓĪŻ
AS┼R┤▓į\öÓ╣▓ūRś╦£╩░³└©Š∙│÷¼F▒Ē¼FŻ©Äū║§╚½▓┐╗╝š▀Š∙│÷¼FĄ─▒Ē¼FŻ®ĪóĮø│Żąį▒Ē¼FŻ©│¼▀^80Żź╗╝š▀│÷¼FĄ─▒Ē¼FŻ®ęį╝░ŽÓĻPąį▒Ē¼FŻ©╔┘ė┌80Żź╗╝š▀│÷¼FĄ─▒Ē¼FŻ®ĪŻŠ∙│÷¼FĄ─▒Ē¼F░³└©░lė²▀t£■ĪóšZčįšŽĄKĪó▀\äė╗“ŲĮ║ŌšŽĄKŻ¼═©│Ż╩Ū▓ĮæBšŽĄK║═Ż©╗“Ż®ų½¾wŅØäėĪóąą×ķ¬Ü╠ž░³└©ŅlĘ▒┤¾ą”/╬󹔯╗├„’@Ą─┐ņśĘ┼eų╣Īóęū┼dŖ^ąįŻ¼═∙═∙░ķėą╩ųŅØ║═▀\äė▀^Č╚ąą×ķĄ╚ĪŻĮø│Żąį▒Ē¼F░³└©Ņ^ć·░lė²čė▀tĪó░d░B░lū„ęį╝░╠žš„ąį«É│Ż─XļŖłDŻ©Ė▀▓©Ę∙╝¼-┬²▓©Ą╚Ż®ĪŻŽÓĻPąį▒Ē¼F░³└©ŲĮšĒ/šĒ╣Ū░╝Ž▌ĪóŽ┬ŅM═╗│÷ĪóīÆūņĪó²X┐pŽĪ╩ĶĪóŅlĘ▒┴„┐┌╦«Īó▀^ČÓĄ─ūņ▓┐äėū„Īó─w╔½╝░░l╔½£\ĄŁĪó▀\äėĢrŪ³Ū·╩ų▒█Īó╦»├▀šŽĄKĄ╚ĪŻ
▓╗═¼░l▓ĪÖCųŲ╗╝š▀į┌į\öÓ▀^│╠ųą▓╔╚ĪĄ─į\öÓ╩ųČ╬▓╗═¼ĪŻ▀zé„īWÖz£y╩ūŽ╚▀xō±Ą─╩ŪDNA╝ū╗∙╗»Ęų╬÷Ż¼▀@ŅÉĘĮĘ©░³└©╝ū╗∙╗»ČÓųž▀BĮėę└┘ć╩Į╠ĮßśöUį÷╝╝ągŻ©MS-MLPAŻ®Īó╝ū╗∙╗»PCRŻ©MS-PCRŻ®Ą╚Ż¼┐╔į\öÓ─Ėį┤15q11.2-q13╚▒╩¦ĪóĖĖį┤å╬ėHČ■¾wŻ©UPDŻ®╗“ėĪėø╚▒Ž▌Ż©IDŻ®╦∙ų┬Ą─╠ņ╩╣ŠC║Žš„Ż¼╔Ž╩÷ĘĮĘ©┐╔į\öÓ80Żźū¾ėęĄ─╗╝š▀ĪŻ╚ń╣¹DNA╝ū╗∙╗»Ęų╬÷ĮY╣¹š²│ŻŻ¼ät┐╔ęį┐╝æ]▀MąąUBE3A╗∙ę“ą“┴ąĘų╬÷Ż¼įōĘĮĘ©┐╔╩╣┤¾╝s10ŻźĄ─╗╝š▀Ą├ĄĮ┤_į\ĪŻ┴Ē═ŌŻ¼╚įėą╝s10Żź╗╝š▀ė╔ė┌╔ą╬┤├„┤_Ą─ų┬▓ĪÖCųŲČ°¤oĘ©Å─Ęųūė▀zé„īW┤_į\Ż¼ų╗─▄ę└ō■Ąõą═▒Ē¼Fū÷│÷┼R┤▓į\öÓĪŻSNP-arrayąŠŲ¼Ęų╬÷┐╔ęįÖz£y15╠¢╚Š╔½¾wĄ─╬ó╚▒╩¦║═UPDŻ¼Ą½¤oĘ©Å─╝╝ąg╔Žģ^äe╠ņ╩╣ŠC║Žš„║═Ųš└ŁĄ┬-═■└¹ŠC║Žš„Ż¼ąĶĮY║Ž┼R┤▓▀Mąąį\öÓŻ¼╗“▓╔ė├MS-MLPAĘĮĘ©▀Mąą“×ūCĪŻ
Ķbäeį\öÓŻ║AS╗╝š▀═©│Ż┤µį┌ĘŪ╠ž«ÉąįŠ½╔±▀\äė░lė²▀t£■║═Ż©╗“Ż®░d░BŻ¼ę“┤╦Ķbäeį\öÓ═∙═∙Š▀ėąĘŪ╠ž«ÉąįĪŻ░³└©─X░cĪóĘŪ▀Mš╣ąį─X▓Ī╗“ŠĆ┴Ż¾w─X╝Ī▓ĪĄ╚Ż¼Ą½╠ņ╩╣ŠC║Žš„Ą─ų½¾wČČäė║═▓╗ĘĆČ©╩ŪŲõ┼c╔Ž╩÷╝▓▓ĪĄ─Ķbäe³cĪŻ
─┐Ū░šJ×ķŻ¼ASąĶę¬┼c╚ńŽ┬╝▓▓Ī▀MąąĶbäeį\öÓŻ║
1.Mowat-WilsonŠC║Žš„
┐╔▒Ē¼F×ķŽ▓┼Ł¤o│ŻĪó░d░B░lū„ĪóŽ┬ŅM═╗│÷ĪóČ·┤╣═╗│÷ĪóčįšZ£p═╦ĪóąĪ─XĪó▒Ń├žŻ¼ėąĢr▀Ć┐╔▒Ē¼F×ķŽ╚╠ņąįŠ▐ĮY─cĪŻŽ╚╠ņąįą─┼K╚▒ōp╗“ļ▌ļš¾w░lė²▓╗╚½ę▓┐╔─▄░l╔·ĪŻMowat-WilsonŠC║Žš„═©│Ż╩Ūė╔ZEB2╗∙ę“ą┬░l═╗ūāī¦ų┬ĪŻ
2.Pitt-HopkinsŠC║Žš„
įō▓Ī╠žš„╩ŪųŪ┴”šŽĄKĪóīÆūņ║═¬Ü╠žĄ─├µ▓┐╠žš„Ż¼ęį╝░ķgą¬ąį▀^Č╚═©ÜŌ░ķ║¶╬³Ģ║═ŻĪŻąĪŅ^╗¹ą╬Īó░d░B░lū„Īó╣▓Ø·╩¦š{▓ĮæB║═┐ņśĘéĆąįĄ╚▒Ē¼F┼cASėąųž»BĪŻ3Üq║¾╗╝š▀┐╔ėą╠žš„ąįĄ─╚šķg▀^Č╚ōQÜŌĪŻįō▓Īų┬▓Ī╗∙ę“×ķTCF4ĪŻ
3.ChristiansonŠC║Žš„
įō▓Ī┼R┤▓╠žš„░³└©├„’@┐ņśĘĄ─ąįĖ±Īóć└ųžĄ─šJų¬čė▀tĪó╣▓Ø·╩¦š{ĪóąĪŅ^╗¹ą╬║═░d░BšŽĄKĪŻ╗╝š▀╔Ē▓─╩▌ąĪŻ¼10Üq║¾┐╔─▄¤oĘ©ąąū▀Ż¼▓┐Ęų╗╝š▀┐╔─▄ėąąĪ─X║═─XĖ╔╬«┐sĪŻ▒M╣▄ā╔š▀Š∙┤µį┌░d░B░lū„Ż¼Ą½─XļŖłD╠ž³cėą▓╗═¼▒Ē¼FŻ║ASėą╠žš„ąįĄ─Ė▀▓©Ę∙╝¼-┬²▓©Ż©1.5Ī½3HzŻ®╠ž³cŻ¼Č°ChristiansonŠC║Žš„Š▀ėą┐ņŻ©10Ī½14HzŻ®Ą─▒│Š░╣Ø┬╔ĪŻChristiansonŠC║Žš„╩Ūė╔SLC9A6╗∙ę“═╗ūāę²ŲĄ─ĪŻ
4.RettŠC║Žš„
Ąõą═▒Ē¼F×ķ│÷╔·║¾įńŲ┌ųŪ┴”▀\äė░lė²š²│ŻŻ¼6Ī½18éĆį┬│÷¼FšJų¬╝░▀\äė╣”─▄Ą─Ą╣═╦Ż¼šZčį╝░╔ńĢ■Į╗═∙─▄┴”Ž┬ĮĄŻ¼Ņ^ć·į÷ķLŠÅ┬²Ż¼å╩╩¦ęč½@Ą├Ą─╩ųĄ─Š½╝Ü╣”─▄Ż¼│÷¼F╩ųĄ─┐╠░Õäėū„ĪŻ│÷¼F¾@ž╩ĪóąĪŅ^╗¹ą╬║═ć└ųžčįšZšŽĄKĄ─┼«ąįAS╗╝š▀ąĶūóęŌ┼cRettŠC║Žš„ĶbäeĪŻRettŠC║Žš„Ą─┼«ąį╗╝š▀═©│Ż▓╗╚▌ęū┐ņśĘŻ¼Č°╗╝ėąASĄ─┼«ąį╗╝š▀ät╚▒Ę”╔±Įø═╦ąąąį▓Ī│╠║═ŅlĘ▒Ą─¤o─┐Ą─ąįĄ─╩ųĄ─äėū„ĪŻį┌─Ļ²g▌^┤¾┼«ąį╗╝š▀ųą▀Mąąā╔š▀Ą─Ķbäeį\öÓĢr┤µį┌ę╗Č©└¦ļyŻ¼ąĶę└┐┐▀zé„īWÖz£y▀Mąą┤_į\ĪŻRettŠC║Žš„╩Ūė╔MECP2╗∙ę“═╗ūāę²ŲĄ─X▀Bµiļ[ąį▀zé„▓ĪĪŻ
ęį╬╣B└¦ļy║═╝ĪÅł┴”Ą═Ž┬×ķų„ę¬▒Ē¼FĄ─AS╗╝š▀ąĶūóęŌ┼cŲš└ŁĄ┬-═■└¹ŠC║Žš„ĶbäeĪŻ┤╦═ŌŻ¼Ųõ╦¹╝▓▓ĪŻ¼╚ń2q23.1╬ó╚▒╩¦Īó22q13.3╚▒╩¦ŠC║Žš„Īó─ąąįMECP2ųžÅ═ĪóŽ┘▄š╦ßń·ńĻ╦ß┴čĮŌ├Ė╚▒Ę”░YĪó┼cĄ═╝ū┴“░▒╦ß║═Ė▀═¼ą═░ļļū░▒╦ßč¬░YŽÓĻPĄ─üå╝ū╗∙╦─Üõ╚~╦ß▀ĆįŁ├ĖŻ©MTHFRŻ®╚▒Ę”░YĪóŽ╚╠ņąį╠Ū╗∙╗»šŽĄKĪóKleefstraŠC║Žš„ĪóHERC2ŽÓĻPĄ─šJų¬šŽĄKĪóWACŽÓĻPĄ─ųŪ┴”šŽĄKĄ╚ęÓąĶū├ŪķĶbäeĪŻ
╚²Īóų╬»¤ĘĮ╩Į
ų╬»¤Ż║įō▓ĪŲ∙Į±╔ą¤o╠žą¦ų╬»¤ĪŻų„ę¬╣żū„į┌ė┌į\öÓ║¾ĮĪ┐Ą╣▄└ĒĪŻ─┐Ū░ų„ę¬╩Ūßśī”┼R┤▓▒Ē¼F▀MąąĘeśOĄžī”░Y╝░ų¦│ųų╬»¤Ż¼ėąų·ė┌╠ßĖ▀AS╗╝ā║Ą─╔·╗Ņ┘|┴┐ĪŻ
«öą┬╔·ā║│÷¼F╬╣B└¦ļyĢrŻ¼ąĶę¬▓╔ė├╠ž╩Ō─╠ūņ║═Ųõ╦¹ĘĮĘ©╣▄└Ē╗╝ā║╦▒╬³─▄┴”╚§╗“▓╗ģfš{Ą─ŪķørŻ╗╩╣ė├┐╣░d░B╦Ä╬’┐žųŲ░d░B░lū„Ż¼─┐Ū░Ģ║¤o═Ų╦]╦Ä╬’Ż╗ī”ė┌│÷¼F╔ńĢ■ŲŲē─ąį╗“ūį╬ęé¹║”Ą╚▓╗┴╝ąą×ķĄ─╗╝ā║Ż¼Į©ūh▓╔ė├ąą×ķ»¤Ę©▀MąąĖ╔ŅAŻ╗æ¬į┌▀m«öĄ─Ģr║“▒Mįń╩╣ė├▌oų·£Ž═©╣żŠ▀Ż¼╚ńłDŲ¼┐©╗“Į╗┴„░ÕŻ¼ęįĖ─╔Ų╗╝ā║čįšZšŽĄKĄ─ŪķørŻ╗└¹ė├ąžč³ūĄŖA┐╦║═Ż©╗“Ż®╩ųągĖ╔ŅAüĒų╬»¤╝╣ų∙é╚═╣░YĀŅĪŻ
ėąę╗ą®╗∙ę“ų╬»¤š²į┌ĘeśOķ_š╣ųąŻ¼▒╚╚ń└¹ė├Č╦┴Ż├ĖęųųŲä®╗“Ę┤┴x╣č║╦▄š╦ß╝ż╗Ņ╝ū╗∙╗»│┴─¼Ą─ĖĖį┤UBE3AĄ╚╬╗╗∙ę“Ż¼ęįŲ┌▀_ĄĮį÷╝ėĖĖį┤UBE3AĄ╚╬╗╗∙ę“Ą─▒Ē▀_Īóča│õ─Ėį┤UBE3AĄ╚╬╗╗∙ę“╚▒Ž▌ĪóūŅĮKų╬»¤╝▓▓ĪĄ──┐Ą─ĪŻ
į\»¤┴„│╠łDŻ║
Angelman╩Ž░Y║“╚║Ż©ASŻ®Ęųūėį\»¤┴„│╠łD
ģó┐╝╬─½IŻ║
[1] Jiang Y, Lev-Lehman E, Bressler J, et al. Genetics of Angelman syndrome.Am J Hum Genet,1999,65:1-6.
[2] Clayton-Smith J, Pembrey ME. Angelman syndrome. J Med Genet,1992,29:412-415.
[3] Dagli A, Buiting K, Williams CA. Molecular and clinical aspects of Angelman syndrome. Mol Syndromol,2012,2(3-5):100-112.
[4] Williams CA, Beaudet al, Clayton-Smith J, et al. Angelman syndrome 2005Ż║updated consensus for diagnostic criteria. Am J Med Genet A,2006,140(5):413-418.
[5] Bai JL, Qu YJ, Jin YW, et al. Molecular and clinical characterization of Angelman syndrome in Chinese patients. Clin Genet,2014,85(3):273-277.
[6] https://www.ncbi.nlm.nih.gov/books/NBK1144/
░µÖÓ┬Ģ├„
ęį╔Žā╚╚▌üĒūį┴╝ßtģR-║▒ęŖ▓Īą┬▀Mš╣Ż¼╚ńėąĮ©ūh╗“ę╔å¢Ż¼ÜgėŁų┬ļŖ18017449015ĪŻ
 ķL░┤Č■ŠS┤a
ķL░┤Č■ŠS┤aĻPūóŠ½▓╩ā╚╚▌





